


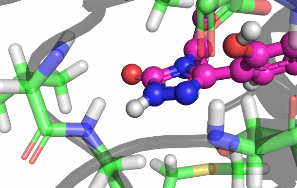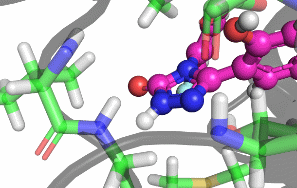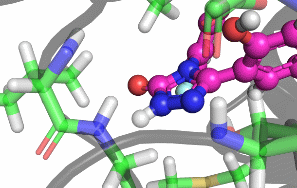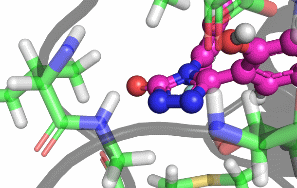
My research focuses on designing and implementing methods to allow the protonation states of certain titratable sites to vary during a classical MD simulation. Typically, the vast majority of biomolecular simulations are setup by first assigning a fixed protonation state both to all the protein residues and also any ligands present. While this assumption is reasonable in most cases, in situations where the pKa of a ligand is near the pH of the total system (typically biological pH), restricting residues and ligands to both a single protonation and tautomeric state can prevent relevant configurations from being sampled. As a result, calculated properties such as binding free energies are likely to be incorrect.There are a number of existing methods that can be used to sample from a semi-grand canonical ensemble, Non-equilibrium Candidate Monte Carlo (NCMC) is the one that I will be applying initially to this problem of dynamic protonation state sampling. Short NCMC switching trajectories can be used to gradually either turn or or turn off a proton before the final configuration is subjected to an acceptance test. This will allow pre-defined titratable sites, primarly on ligands and certain residues (e.g. histidine), to potentially change their protonation state at regular intervals.